
05多位点序列分析(MLST)分析
多位点序列分析(MLST)分析
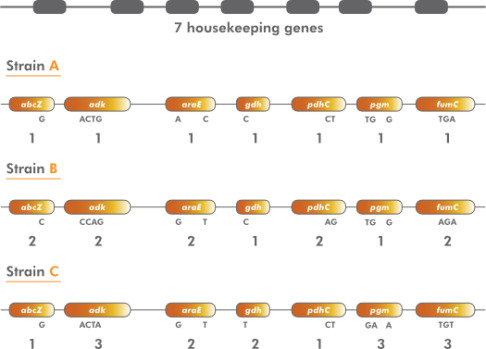
多位点序列分型的原理:MLST方法一般测定6~10个管家基因内部400~600bp的核苷酸序列,每个位点的序列根据其发现的时间顺序赋予一个等位基因编号,每一株菌的等位基因编号按照指定的顺序排列就是它的等位基因谱,也就是这株菌的序列型(sequence type,ST)。这样得到的每个ST均代表一组单独的核苷酸序列信息。通过比较ST可以发现菌株的相关性,即密切相关菌株具有相同的ST或仅有极个别基因位点不同的ST,而不相关菌株的ST至少有3个或3个以上基因位点不同。
多位点序列分析(MLST)分析
多位点序列分型(MLST)是一种分子分型技术,通过该技术,通常会对部分精心挑选的管家基因(位点)进行测序。
在典型的MLST方法中,重组的发生频率要比点突变高得多。因此,人们不会研究菌株之间的总的序列相似性。相反,而去筛选给定位点的每个序列与该位点的已知序列的相似性。如果序列不同,则将其视为新的等位基因,并为其分配唯一的(任意)等位基因编号。如果研究了七个管家基因,那么每个菌株的特征就是七个等位基因的图谱。等位基因图谱可被认为是由7个分类字符组成的字符集。MLST已成功用于研究种群遗传学和流行细菌和其他微生物的重建微观进化。
Bionumerics中的MLST分析
Applied Maths公司通过使用最小生成树为MLST数据的分析做出了贡献(请参阅L. Vauterin和P. Vauterin.综合数据库和分析。在E. Stackebrandt,ED,分子鉴定,系统学和原核生物的种群结构中。Springer-Verlag Berlin Heidelberg,2006年,以及许多研究文章)。MLST插件的使用,使BioNumerics软件被广泛用于MLST序列的存储和分析。BioNumerics会自动分析一批序列跟踪文件,连接到在线MLST数据库,检索相应的等位基因编号,序列类型以及可用的克隆复合体信息。 BioNumerics可以在几秒钟内处理数百个分离株。结果被存储在数据库中,并可使用BioNumerics非常好用的分析工具进行统计和种群分析、聚类、分组、鉴定。
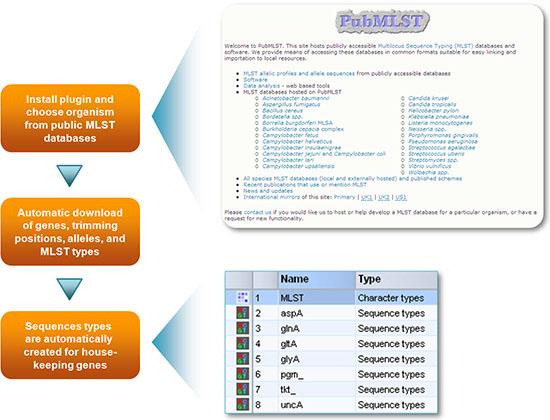
MLST数据库的简易配置
MLST数据库可以轻松设置,例如大肠杆菌、肠炎沙门氏菌、肺炎克雷伯菌、弯曲杆菌、鲍曼不动杆菌、铜绿假单胞菌或可从在线存储库(例如mlst.net或pubmlst.org网站)获得的任何其他生物,只需从列表中选择生物即可。BioNumerics可以在本地下载等位基因,剪切位置和MLST类型,并在启动时更新其数据库,或从Web服务获取每次分析的数据。或者,用户可以设置自己的MLST框架。
配置框架
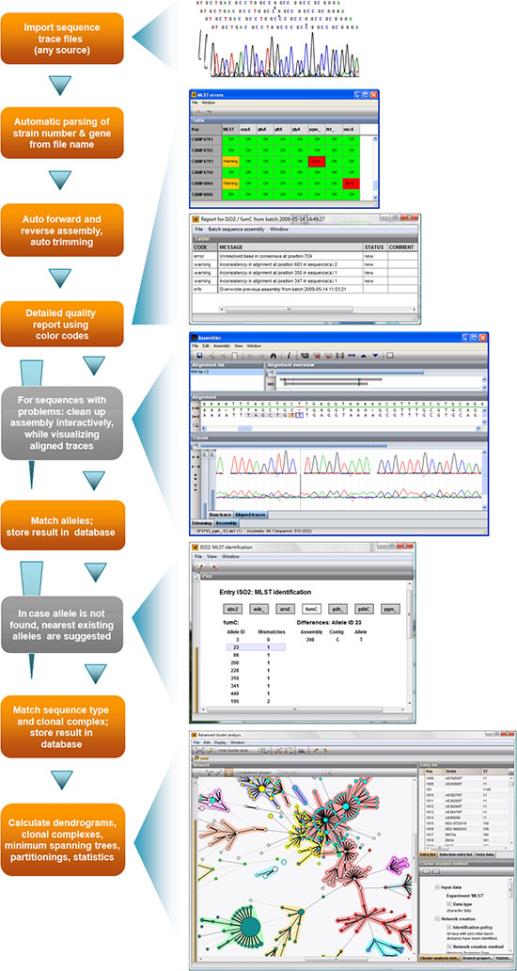
全自动的处理流程:
l 自动从各种来源(AB、Beckman、Amersham、FASTA)导入和拼接一批测序跟踪文件;使用解析规则将文件名解析为菌株和基因信息。
l 使用开始和停止标记自动剪切一致性序列,并按正确的方向放置。
l 序列拼接完成后,将显示预览报告,列出每种菌株/基因组合的状态。
l 双击有问题的contig以显示详细信息窗口。
l 双击一个特定的问题以打开选择问题位置的拼接窗口。
l 对于每个问题位置,显示最接近的现有等位基因和建议的碱基-通过谱图易于验证。
l 可以通过与MLST服务器数据库的实时连接或与本地存储的等位基因数据库进行比较(更快)来识别等位基因和MLST类型。在后一种情况下,本地数据库可以在启动时自动更新。
l 自身菌株的等位基因和MLST类型信息存储在数据库中,可以随时更新以选择菌株。 BioNumerics将提示在MLST服务器数据库中发生的等位基因/ MLST类型定义的任何更改。
l 使用标准或自定义优先级规则以及分支重要性支持指示,在目前可用的最精细和最全面的聚类分析应用程序中计算种群建模网络。
l 计算和显示克隆复合体的分组,并使用BioNumerics丰富的统计工具。
工作流程:


中国区官方授权经销商
以上资料由BioNumerics中国区官方授权经销商独家提供,如需转载请主动标明出处。
客服专线:010-53639817 53639871
市场专线:15712982811 13552183571
QQ客服:3545869936 3316774663
搜索关键词:
比利时Applied-Maths Bionumerics、比利时Bionumerics、BioNumerics生物信息分析软件、BioNumerics生物分析软件、BN分析软件、PFGE分析软件、Bionumerics软件7.6、Bionumerics软件6.6、食源性疾病、