
SarsCoV2插件
8.2 导出accession 号至BLAST Entrez
1. 引言
手册中主要介绍了对SARS-CoV-2基因组序列的处理和分析,序列可以是从公共数据库下载或者是本地生成的数据。每个单独基因组序列被分成一系列子序列(从序列中提取多个部分序列),每一个序列会根据参考基因组进行SNP的分析。所有的SNP以开放的(动态)字符集存储,这些字符集能够在现有的最高分辨率下进行简单的对比和菌株分型。
SarsCoV2插件是免费提供的。如果要安装该插件,Bionumerics软件的最低配置为“特征数据模块”、“序列数据模块”、“树状网络分析模块”和“基因组分析工具模块”。
确保Bionumerics软件已是最新版本(https://www.applied-maths.com/download/software),安装手册可从https://www.applied-maths.com/download/manuals下载。
双击电脑上Bionumerics软件的程序图标进入到软件的启动窗口(图2.1)。
通过点击![]() 按钮创建新的数据库,点击
按钮创建新的数据库,点击![]() 按钮或者双击数据库列表中的名称可以打开已创建的数据库。
按钮或者双击数据库列表中的名称可以打开已创建的数据库。
2.1点击Bionumerics软件启动窗口中的![]() 图标进入新数据库的向导。
图标进入新数据库的向导。
2.2为数据库命名(例如My database),然后点击<Next>。
弹出的新对话框会提示数据库类型。
2.3保持默认选项“Create new”然后点击<Next>。
弹出的新对话框会提示数据库引擎。
2.4保持默认选项(图2.2)然后点击<Finish>完成新数据库的设置。
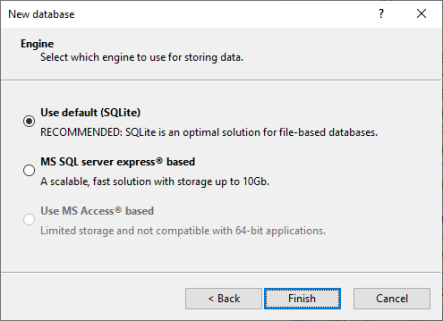
图2.2 选择数据库引擎
弹出插件对话框,允许您安装其他功能。
2.5点击<Proceed>进入数据库主界面。
在安装SarsCoV2插件(图2.4)前,请确定您已安装Sequence extraction插件:
3.1点击数据库主界面左上角 File > Install / remove plugins.. 调用插件对话框。
3.2在插件对话框中点击 Utilities 子菜单,从列表中选择Sequence extraction并点击<Activate>按钮。
3.3确认安装插件(图2.3)。
插件成功安装后,对话框中会标记为绿色对勾(图2.4)。
3.4关闭插件对话框。
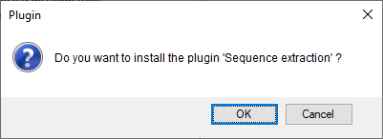
图2.3 确认安装插件
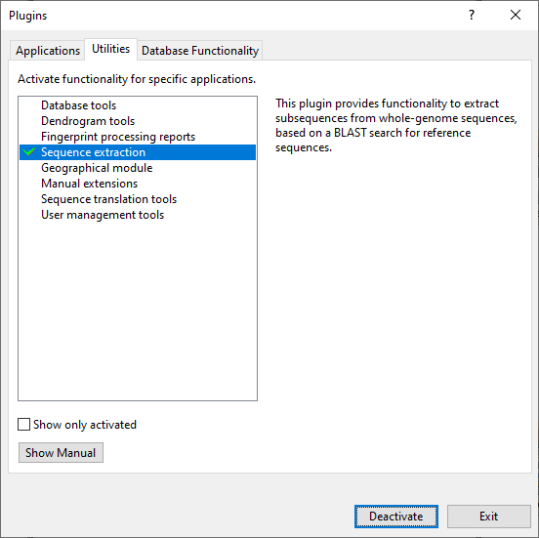
图2.4 安装插件
4.1点击数据库主界面左上角 File > Install / remove plugins... 调用插件对话框。
4.2在插件对话框中点击 Database Functionality 子菜单,然后点击<Add / Update...>按钮。
4.3点击<Browse>按钮,选择提供的SarsCoV2Client.BPL文件(图2.5)。
4.4点击<OK>安装插件。
4.5确认安装插件(图2.6)
弹出Create database components对话框显示了所有插件需要的数据库组件:entry fields,character type实验,sequence type实验(图2.7)。如果有需要的话,这些名称都可以更改。
4.6点击<OK>确认创建数据库组件。
弹出的信息显示插件已安装成功(图2.8)。
4.7点击<OK>。
插件对话框中SarsCoV2插件被绿色对勾标记(图2.9)。
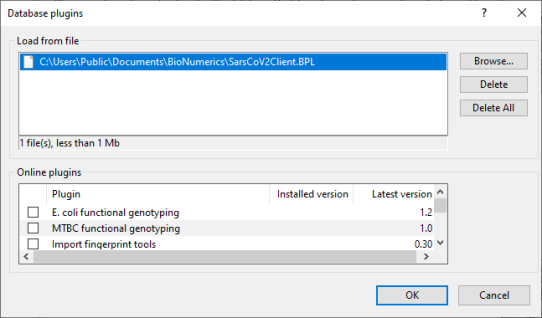
图2.5 浏览选择bpl文件
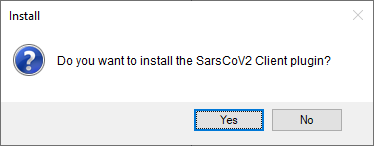
图2.6 确认安装插件
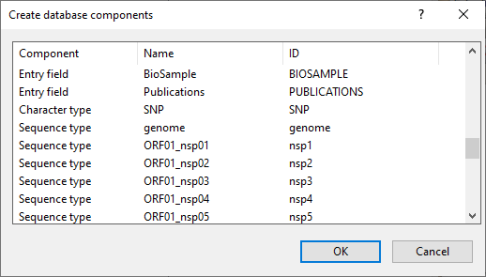
图2.7 新的数据库组件
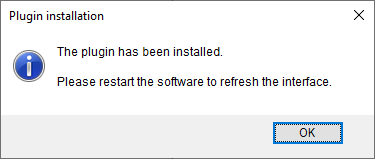
图2.8 插件安装完成
4.8关闭插件对话框
4.9关闭数据库并重新打开来激活SarsCoV2插件的功能
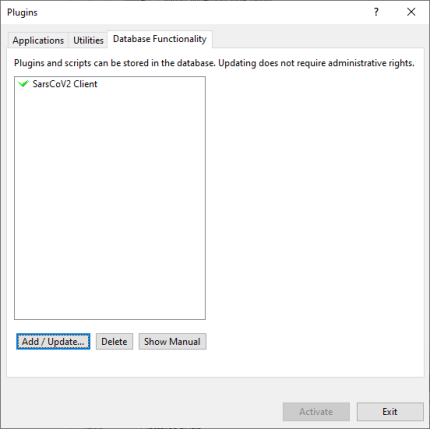
图2.9 插件安装完成
数据库主界面如图2.10所示。
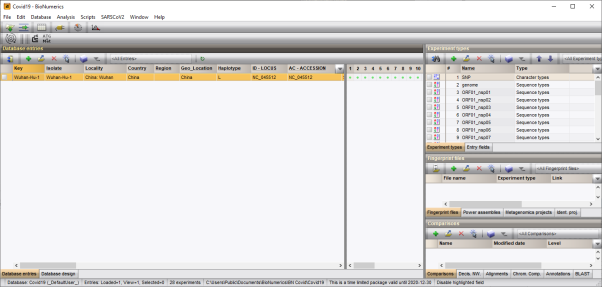
图2.10 安装完SarsCoV2插件后的数据库主界面
安装SarsCoV2插件后数据库会增加SARSCoV2菜单项(图2.11)和以下组件:
l 名为genome的序列类型实验,存储拼接后的全基因组。
l 26个序列类型,存储所提取的子序列。
l 名为SNP的特征类型实验,存储SNP信息。
l 33个信息字段,由标准GenBank元数据字段和NCBI的SARS-CoV-2数据中心列组成。
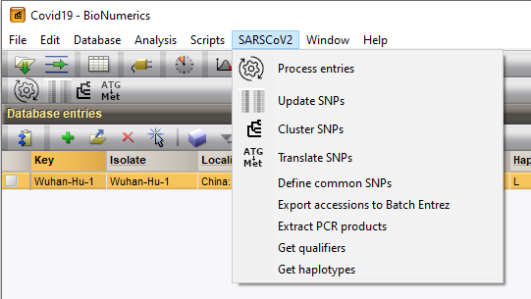
图2.11 菜单项
数据库有一条key为Wuhan-Hu-1的条目。NCBI上SARS-CoV-2的参考序列NC_045512被存储在这一条目的genome序列类型实验中。
4.10点击数据库主界面实验展示面板中对应genome序列类型实验的绿点(默认配置下2号实验),打开序列编辑窗口。
窗口上部分是序列,下部分是序列的图形化展示(图2.12)。Annotation面板显示了NCBI的特征区域,Header面板显示了header信息。
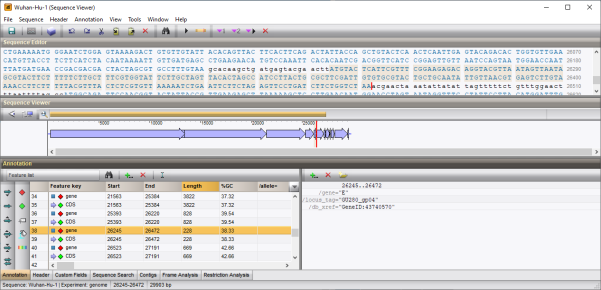
图2.12 序列编辑窗口
4.11关闭序列编辑窗口
SARS-CoV参考序列NC_045512的子序列分别存储在对应的序列类型实验中。序列实验名称为ORF(Open Reading Frame开放阅读框架)后接数字以及可选的nsp(Nuclear Shuttle Protein核穿梭蛋白)组成。例如ORF01_nsp01。这些子序列作为样品序列筛选时BLAST的参考序列(图4.1)。
0.1选择File > Import... 调用数据导入对话框。
Bionumerics软件中所有导入(拼接后)基因组序列的途径都是从Sequence data菜单中。
0.2通过点击Sequence type data旁的“+”标志显示所有序列导入途径(图3.1)。
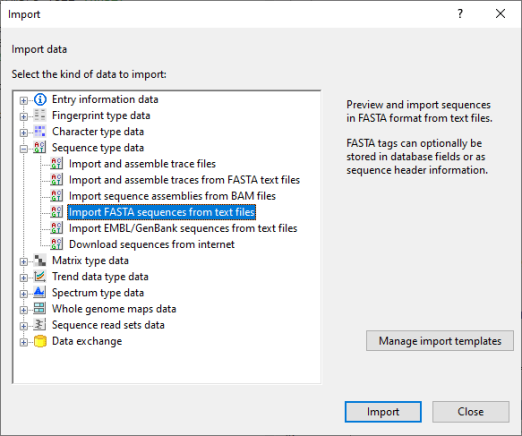
图3.1 数据导入选项
例如,我们会从EMBL/NCBI抓取序列。更多关于其他序列导入方法的详细信息可见网站中的序列手册。
0.3数据导入对话框选择<Manage import templates>。
0.4选择<Import from file>,浏览找到SarsCoV2 template.xml文件,和插件文件放在一起,然后点击<OK>(图3.2)。
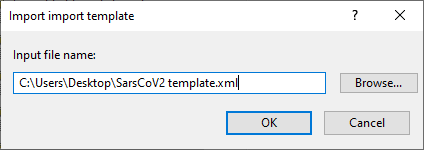
图3.2 XML模板
导入模板将EMBL/NCBI标签和SarsCoV2插件创建的条目字段关联。
0.5点击<OK>将导入模板添加到数据库中。
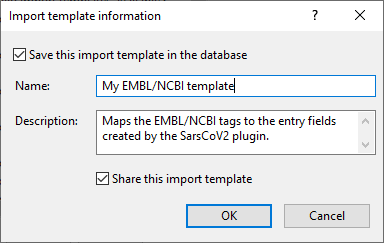
图3.3 导入xml模板
0.6在数据导入对话框中,选择Sequence type data下的 Download sequences from internet并点击<Import>。
0.7在Accession codes输入字段中输入accession号(例如MT385458,MT385436,MT385431),这些号由“,”分隔。
0.8指定“,”作为Separation character,并选择可用的下载站点比如EBI。
0.9勾选 Preview sequences并点击<Next>。
该导入方法会抓取所选数据库的序列并在下一步骤中显示详细信息(图3.4)。
0.10点击<Next>。
导入向导的下一步列出了数据库中导入序列信息的模板。前面步骤导入的预先设置的模板则被列出(图3.6)。
0.11确保选择了My EMBL/NCBI template,并点击<Preview>按钮来检查映射(图3.5)。
0.12关闭预览窗口。
0.13确保选择了My EMBL/NCBI template和genome实验,然后点击<Next>。
0.14点击<Finish>。
条目则被创建并自动是选择状态。条目字段被更新并且序列存储在genome实验中(图3.7)。
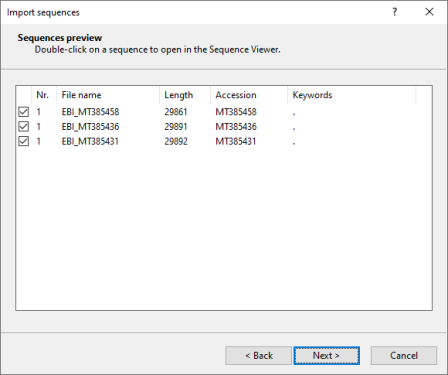
图3.4 抓取的信息
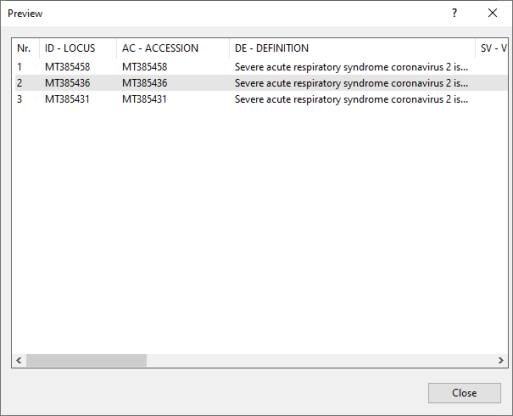
图3.5 预览
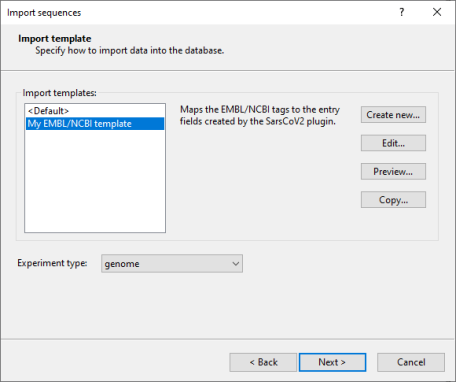
图3.6 导入模板
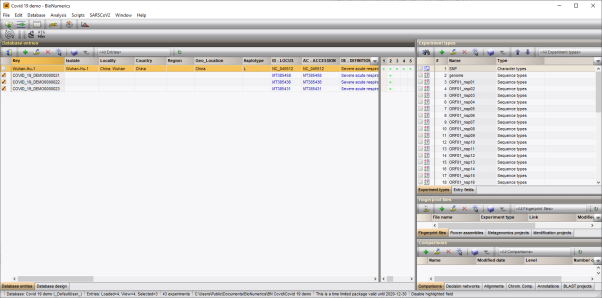
图3.7 导入基因组序列后的数据库主界面
导入并存储在genome实验中的序列现在可以通过SarsCoV2插件进行分析:
0.1数据库的Database entries面板中所选的条目,可以通过Ctrl键选择。同样可以勾选条目旁边的复选框进行选择。
0.2选择菜单栏中SARSCoV2 > Process Entries或者![]() 图标开始处理。
图标开始处理。
包括以下过程:
1. 从genome实验中存储的基因组序列提取26个子序列,并将子序列存储到对应的实验类型中(图4.1)。
2. 通过将一些常见的错义SNP翻译为氨基酸来确定Haplotype(图4.2)。
3. 从带注释的源序列中提取Locality。
SarsCoV2插件通过BLAST方法从genome实验中的序列提取子序列。Wuhan-Hu-1条目的子序列作为BLAST搜索的参考序列。
所选条目的基因组序列找到的子序列被存储在对应的序列类型中。序列实验名称为ORF(Open Reading Frame开放阅读框架)后接数字以及可选的nsp(Nuclear Shuttle Protein核穿梭蛋白)组成。例如ORF01_nsp01。
经过BLAST筛选后,弹出的信息框会询问是否显示BLAST结果的报告(图4.1)。
1.1点击<Yes>打开报告窗口。
报告窗口包含了每个条目的报告(图4.2)。Entries面板中是所有条目的分组。
1.2选择Entries面板中其中一个条目。
所选条目的结果展示在Report面板中,包括处理数据的日期和条目的名称。
图4.1 确认对话框
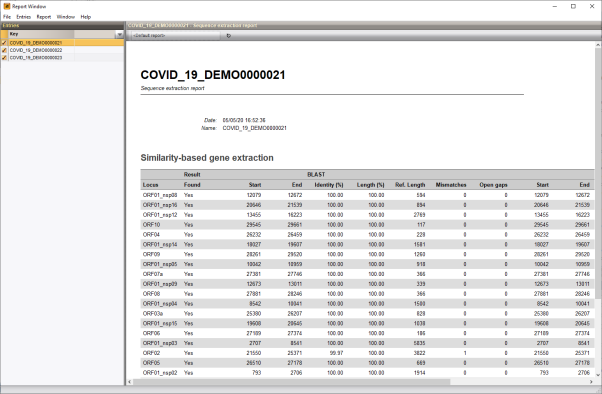
图4.2 报告窗口
对于每个序列类型(Locus列),都表明了是否找到了BLAST hit、所筛选基因序列的起始位置、序列准确度(Identity (%))、序列重叠(Length (%))。
此外,报告了检索到的子序列的长度(Ref length)、参考序列错配的数目(Mismatches)、gaps的数目(Open gaps)和长度修正(如果应用的话)。
1.3关闭报告窗口。
1.4点击其中一个条目的ORF序列实验对应的绿点。
这样会在序列编辑窗口中显示所提取的序列(图4.3为例)。
1.5关闭序列编辑窗口。
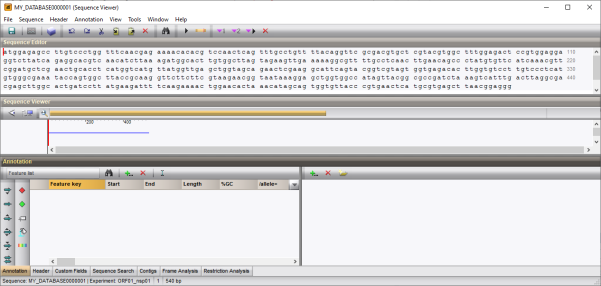
图4.3 ORF01 nsp01序列
在第二步的处理过程,通过将一些常见的错义SNP翻译为氨基酸来确定Haplotype。氨基酸按时间顺序分类,最早的在左边,最近的在右边。Haplotype条目字段显示了Haplotype的结果(图4.4)。
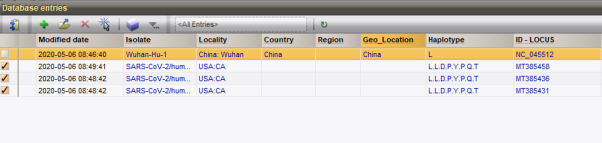
图4.4 确定Haplotype
通过菜单栏的SARSCoV2 > Get haplotypes也能够确定Haplotypes。
最后一步处理过程,元数据通过序列注释被解析(如果有的话),并存储在Isolate和Locality条目字段中。通过菜单栏中SARSCoV2 > Get qualifiers也能获取同样的信息。
通过Bionumerics中的calculated field选项,Locality条目字段(China:Wuhan 或者 USA:CA)可以解析成仅包含国家信息(如China和USA)。相关操作可以在参考手册中找到。
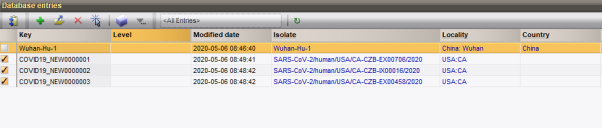
图4.5 从序列注释中提取元数据
5. 计算SNP
提取完子序列后,这些子序列可以进行SNP筛选:
0.1选择数据库主界面中Database entries面板中的条目
0.2选择SARSCoV2 > Update SNPs或者点击![]() 按钮
按钮
子序列则会通过ionumerics内置的SNP分析工具进行SNP筛选(也可以通过菜单栏中的Analysis > Sequence types > Start SNP analysis)。
SNP的结果会基于Relaxed SNP filtering模板进行过滤,过滤后的SNP会存储在SNP特征类型实验中。
Relaxed SNP filtering模板下,非ACGT的碱基也会包含在分析中。然而,非ACGT的碱基实际上不会存储在SNP特征类型实验中,而是用缺失值代替。
经过SNP筛选后,弹出的信息对话框会显示检测的SNP的数目(图5.1)。如果检测到新的SNP位置,同样会显示出并且这些新的SNP位置会自动添加到SNP字符集中。
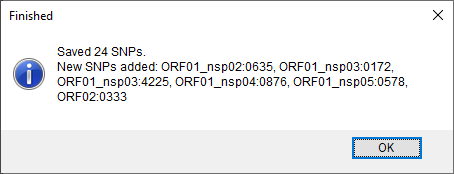
图5.1 SNP信息
0.3点击<OK>关闭确认窗口。
0.4点击其中一个条目在实验数据展示面板中对应SNP特征实验的绿点,打开特征实验卡。
特征实验卡列出来样品中所有检测到的SNP。Mapping列显示了碱基(图5.2)。
0.5点击实验卡左上角关闭实验卡。
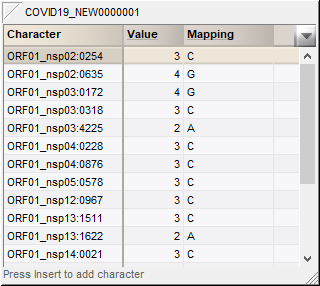
图5.2 SNP特征信息卡
6. SNP数据的聚类
0.1在数据库主界面的Database entries面板中选择想要聚类的条目。
0.2选择SARSCoV2 > Cluster SNPs或者点击主界面![]() 按钮对选择条目进行聚类。
按钮对选择条目进行聚类。
在第一步中,所选的条目都要经过数据处理步骤的提取序列才能进行筛选。
对于子序列有缺失、SNP特征信息中不完整的条目会从聚类比较中排除。警告信息的对话框会提示用户(图6.1)。
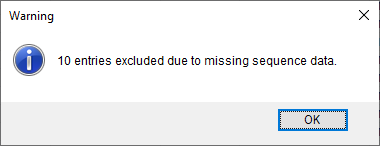
图6.1 从分析中排除的条目
比较窗口出现,如图6.2:
l 基于SNP实验类型计算相似矩阵,使用Categorical (differences)相似系数,显示在Similarities面板中。
l 基于Complete linkage算法得到树状图,显示在Dendrogram面板中。
l 在Experiment data面板中,只显示SNP字符集中的多态SNP。
0.3计算树状图的参数设置可以通过Clustering > Show information调用。
0.4想要显示节点上SNP差异的数目,可以选择Clustering > Dendrogram display settings...,并勾选Show node information。
在比较窗口中,可以根据数据库字段进行分组(比如基于地理位置或者haplotype),或者其他字段。
0.5想要基于数据库字段创建分组,只需右键点击Information fields面板中的字段名称,并选择Create groups from database field。想要基于选择的条目自定义分组,只需使用比较窗口中Groups菜单栏中的命令。
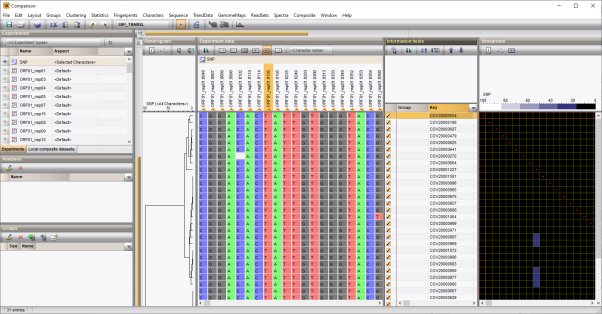
图6.2 比较窗口
在Advanced cluster analysis窗口中可以计算最小生成树。可以在比较窗口中以下操作实现:
0.6确保在比较窗口中的Experiments面板选择了SNP的实验。
0.7 选择Clustering > Calculate > Advanced cluster analysis...或点击![]() 图标 , 并选择Advanced cluster analysis执行创建网络向导。
图标 , 并选择Advanced cluster analysis执行创建网络向导。
0.8选择MST for categorical data,然后点击<Next>。
最小生成树则被计算并显示在Advanced cluster analysis窗口中(图6.3)。
0.9关闭Advanced cluster analysis窗口。
0.10通过File > Save as...保存该次对比,并通过File > Exit关闭对比。
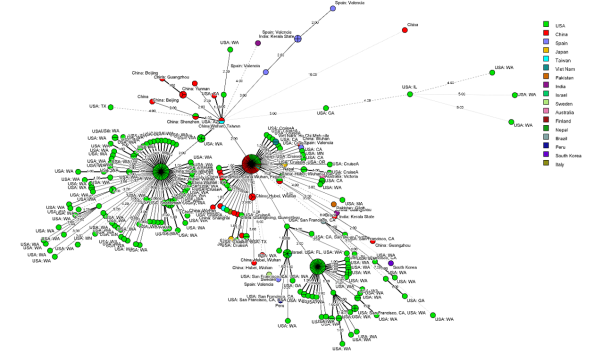
图6.3 定义分组后的最小生成树
7. 翻译SNPs
通过数据库主界面的SARSCoV2 > Translate SNPs,存储在SNP实验的SNP将进行翻译。
0.1选择SNP实验有数据的条目。
0.2选择SARSCoV2 > Translate SNPs或者点击![]() 按钮。
按钮。
第一次进行上述操作时,SNP TRANSL实验将被创建并添加到Experiment types面板中。
所选条目的SNP实验中的SNP进行翻译,并且氨基酸信息存储到SNP TRANSL实验中。
0.3点击对应SNP TRANSL实验数据的绿点打开特征实验卡。
氨基酸信息在Mapping列中显示(图7.1)。
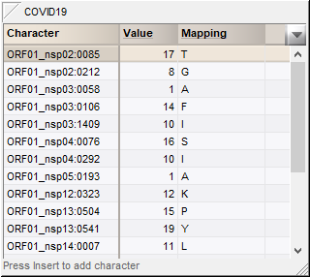
图7.1 特征信息卡
0.4 点击特征信息卡左上角关闭。
该翻译工具假设每个序列的框架都从位置1开始。
通过SarsCoV2插件,PCR产物能够基于WHO标准的引物序列提取(https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/laboratory-guidance)。
1.1在数据库主界面选择想要出PCR产物的条目。
1.2选择SARSCoV2 > Exctract PCR products。
第一次上述操作会创建新的实验类型。
提取的PCR产物存储在对应的PCR序列实验中(图8.1)。
图8.1 存储PCR产物的序列类型
在比较窗口中,可以进一步对指定PCR产物进行分析:
1.3选择想要分析的条目。
1.4使主界面右下方的Comparisons面板处于高亮状态,然后选择主界面菜单栏的Edit > Create new object...对所选条目创建对比。
1.5点击Experiments面板中序列类型实验旁边的![]() 图标(图8.2)。
图标(图8.2)。
可以使用Bionumerics中的序列分析工具进一步分析序列。
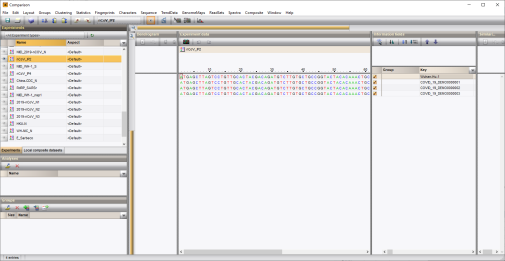
图8.2 PCR产物
8.2 导出accession 号至BLAST Entrez
Bionumerics软件中的数据导入对话框已有标准的导入工具,从NCBI下载GenBank序列,但是不适用于新序列的导入。
要批量检索GenBank格式的序列,请按照以下步骤操作:
2.1选择要导出accession号的条目。
2.2选择SARSCoV2 > Export accessions to Batch Entrez。
2.3浏览选择一个已有的文件夹然后点击<OK>(图8.3)。

图8.3 浏览选择文件夹
该命令将accession号(存储在AC – ACCESSION字段)导出至所选文件夹中以空格分隔的文本文件,然后浏览器打开NCBI BLAST Entrez网站(图8.4),从网站中可以选择accessions的文件(通过<Browse>按钮)来检索GenBank格式的序列。
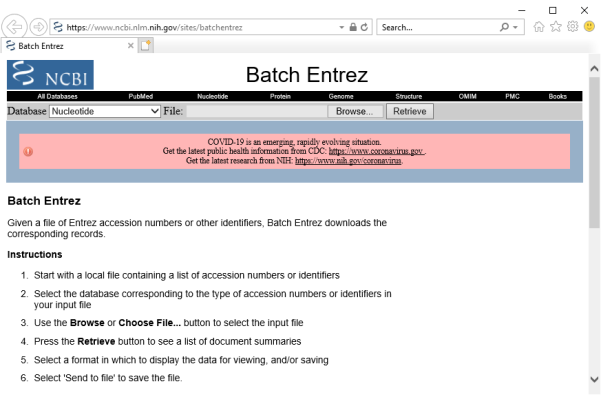
图8.4 Batch Entrez
3.1数据主界面选择想要包含在SNP筛选中的条目。
3.2选择SARSCoV2 > Define common SNPs。
3.3对话框中指定最小频率(图8.5)并点击<OK>。
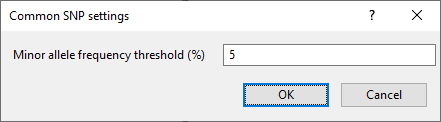
图8.5 指定阈值
基于提供的频率阈值,被鉴定的相同SNP显示出来(图8.6)。
3.4点击<OK>关闭对话框。
相同SNP保存至SNP实验中的Common视图中:
3.5双击主界面Experiment types面板中SNP实验打开特征类型窗口(图8.7)。
3.6点击工具栏中的下拉框,从列表中选择Common特征视图
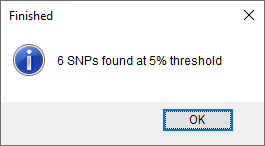
图8.6 结果
根据命令SARSCoV2 > Define common SNPs,相同SNP被鉴定并显示出来。
3.7关闭特征类型窗口。
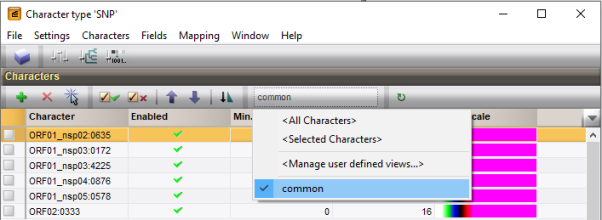
图8.7 特征视图