

让人印象深刻的分析工具集
基于WGS的数据分析工具集
基于wgMLST的高分辨率打字的全自动管道
来自特定于有机体的公共参考数据库(例如BIGDSdb)的wgMLST命名和子类型计划的同步
可以灵活选择键入基因座(例如cgMLST,MLST)或任何其他用户定义的选择
基于原始测序数据(无装配)和从头装配重叠群(基于装配)的wgMLST等位基因分配
对管道中不同步骤的深入质量评估
与BIONUMERICS计算引擎集成,可进行高通量分析
42个内部开发的细菌特定方案:
| Acinetobacter baumannii | Enterobacter cloacae | Lactobacillus sanfranciscensis | Neisseria gonorrhoeae | Staphylococcus pseudointermedius |
| Bacillus cereus | Enterococcus faecalis | Legionella pneumophila | Neisseria meningitidis | Stenotrophomonas maltophilia |
| Bacillus subtilis | Enterococcus faecium | Leuconostoc spp. | Pasteurella multocida | Streptococcus agalactiae |
| Burkholderia cepacia complex | Enterococcus raffinosus | Listeria monocytogenes | Proteus vulgaris | Streptococcus mitis/oralis |
| Brucella spp. | Escherichia coli / Shigella | Micrococcus spp. | Pseudomonas aeruginosa | Streptococcus pyogenes |
| Campylobacter coli - C. jejuni | Francisella tularensis | Mycobacterium bovis | Salmonella enterica | Weissella spp. |
| Citrobacter spp. | Klebsiella aerogenes | Mycobacterium kansasii | Serratia marcescens | |
| Clostridioides difficile | Klebsiella oxytoca | Mycobacterium leprae | Staphylococcus aureus | |
| Cronobacter spp. | Klebsiella pneumoniae | Mycobacterium tuberculosis | Staphylococcus epidermidis |
-
内部开发的wgSNP分析流程,从原始读取开始
-
灵活的基因组定位到多个参考基因组
-
使用Bowtie2 *或SNAP **等参考算法之一执行读取映射
-
提供各种SNP过滤模板
-
可以根据覆盖率或质量标准,位置,突变类型等灵活地创建自己的SNP过滤模板
-
保留的SNP的深入质量评估
-
与BIONUMERICS计算引擎集成,可进行高通量分析
-
集成的CFSAN SNP管道***(美国食品和药物管理局食品安全和应用营养中心),
可以从NGS数据中生成SNP矩阵,用于通常与食品安全相关的病原生物的系统发育分析。
* Langdon, William B. "Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks." BioData mining 8.1 (2015): 1.
** Zaharia, Matei, et al. "Faster and more accurate sequence alignment with SNAP." arXiv preprint arXiv:1111.5572 (2011).
*** Davis, Steve, et al. "CFSAN SNP Pipeline: an automated method for constructing SNP matrices from next-generation sequence data." PeerJ Computer Science 1 (2015): e20.
-
比较基因组图谱和点图表示法,以正向或反向表示同源序列
-
多种基因组比对功能
-
基因组聚类和系统发育分析
-
基于基因组比对的SNP分析和dN / dS计算
-
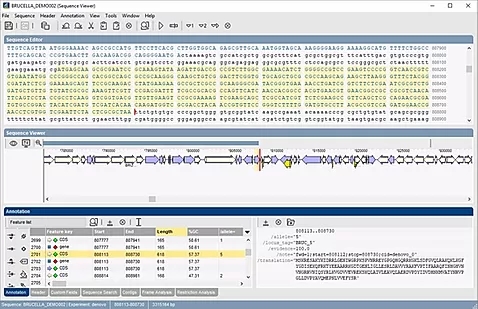
鉴定原核基因组中的编码区
-
基于特征同一性和染色体同义针对一个或多个参考序列的序列注释
-
使用集成的Prokka *管线对序列进行注释
-
与BIONUMERICS计算引擎集成,可进行高通量分析
* Seemann, Torsten. "Prokka: rapid prokaryotic genome annotation." Bioinformatics 30.14 (2014): 2068-2069.
*Zerbino, Daniel R., and Ewan Birney. "Velvet: algorithms for de novo short read assembly using de Bruijn graphs." Genome research 18.5 (2008): 821-829.
** Bankevich, Anton, et al. "SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing." Journal of computational biology 19.5 (2012): 455-477.
*** Souvorov, Alexandre, Richa Agarwala, and David J. Lipman. "SKESA: strategic k-mer extension for scrupulous assemblies." Genome biology 19.1 (2018): 153.
**** Wick, Ryan R., et al. "Unicycler: resolving bacterial genome assemblies from short and long sequencing reads." PLoS computational biology 13.6 (2017): e1005595.
-
从头汇编,基于集成的短头从头汇编器之一,包括Velvet *,SPAdes **,SKESA ***和Unicycler ****
-
利用Unicycler ****生成更准确的“混合”程序集,同时利用两种数据类型的优势,即短读的准确性和长读的结构解析能力
-
与BIONUMERICS计算引擎集成,可进行高通量分析
-
基于最大简约和最大似然方法估计进化关系,并通过系统发生距离缩放校正,例如Jukes&Cantor或Kimura2
-
通过使用嵌入式标准工具RAxML *或FastTree **,基于最大似然来推断复杂程度不同的系统发育
-
与BIONUMERICS计算引擎集成,可进行高通量分析
*Stamatakis, Alexandros. "RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models." Bioinformatics 22.21 (2006): 2688-2690.
** Price, Morgan N., Paramvir S. Dehal, and Adam P. Arkin. "FastTree 2–approximately maximum-likelihood trees for large alignments." PloS one 5.3 (2010).